密度汎関数法による化学反応解析
本来は観測できない遷移状態の構造も、分子軌道計算によって求められる。
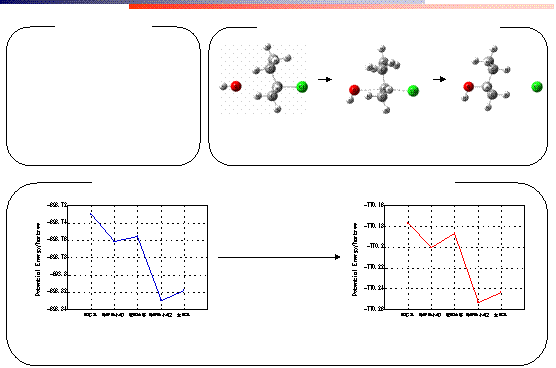
遷移状態
(S)-2-クロロブタン →
(R)-2-ブタノールの SN2 反応
密度汎関数法とは
従来の分子軌道法に代わって、近年注目を集めている理論。Density
Functional Theory を略して DFT とも呼ばれる。 これは Hartree-Fock 法に比べて、分子の化学的特性の算出に優れており、分子の構造や化学値を高精度に、かつ低コストで算出する事が可能である。
(S)-2-クロロブタン →
(R)-2-ブタノールの SN2
反応のポテンシャルエネルギー
局所最小値 1〜遷移状態間の活性化エネルギー
3.72 kcal/mol
溶媒として水 1
分子を付加
局所最小値 1〜遷移状態間の活性化エネルギー
8.38 kcal/mol