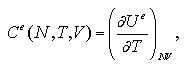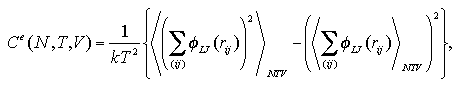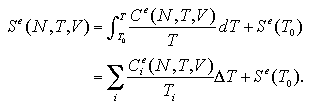Chapter 2:
SUMMARY OF MONTE CARLO SIMULATION
|
|
2.1. Lennard-Jones Potential Function[9] |
We applied the Lennard-Jones (LJ) 12-6 type potential function
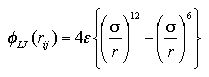 | (2.1) |
for the target particle. The LJ function describes the interaction between two particles as a simple function of which center-center distance, and has been widely used as a function that indicates the intermolecular interaction of monatomic molecules, simplified polyatomic molecules or groups, moreover the intramolecular interaction of the atoms or groups in polyatomic molecules. The symbols ε and σ are the LJ parameters that correspond to the strength of the interaction and the size of the particle. In this study, we used the LJ parameters as the reduced units: ε for energy and σ for length; volume σ3, temperature ε/k, pressure ε/σ3, etc.
|
|
2.2. Model of Homogeneous Nucleation |
In this study we suggest a simple model to estimate the free energy of the homogeneous nucleation in the supersaturated LJ system. Figure 2.2 gives a summary of the model. In the upper figure, a conventional large-system model contains large number, at least several thousands, of target particles. The density corresponds to the supersaturated vapor, and according to circumstances, carrier gas particles are included in the system. During the MC/MD simulations, the small-size nuclei have generated and grown spontaneously in the system. On the other hand, in the lower figure, we focus a specific number of particles N, and put the N target particles and no carrier gas particles into the system. Here, the two extremes are supposed for the N-particle system: namely, cluster phase and monomer phase. The former is a state in which all particles of the system are connecting together as a N-particle cluster, and in the latter all particles are scattering as a supersaturated vapor phase. When the free energies of the cluster phase Gc and the monomer phase Gm are obtained independently, the free energy of the N-particle nucleation ΔG can be assumed as the difference of the latter from the former:
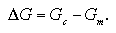 | (2.2) |
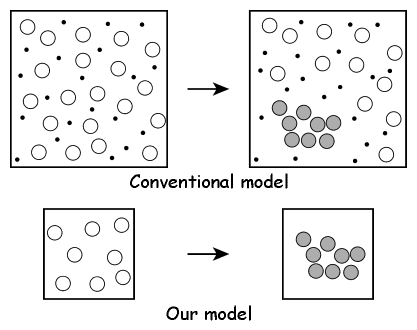 |
| Fig. 2.2. The process of N-particle nucleation in the large system that includes the carrier gas particles is modeled as the nucleation of the whole particles in the N-particle system. The circles indicate the target gas particles, and the shadowed circles the nucleated particles. Though the dots indicate the carrier gas particles in the conventional model, it is not included in our model. Since the cluster phase requires that the all particles in the system consist a unity cluster, the surrounding of the cluster is a vacuum. |
|
|
2.3. Monte Carlo Procedure |
We explain below the detailed procedure of the MC simulations. The volume per particle V/N is chosen as the supersaturated vapor phase. The number of particles N, for which we focused respective values, is set among 2 up to 80. The basic cell is cubic, that contains N target particles and no carrier gas particles, in which the periodic boundary conditions for all three dimensions are adopted. The cut-off distance of the LJ interaction is a half of cell width, and a correction term is adopted for contributions from more distant particles.
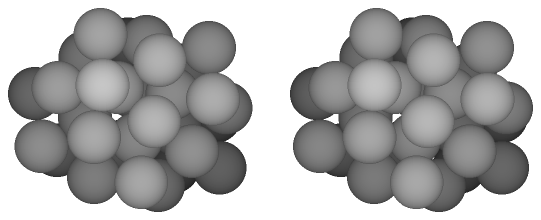
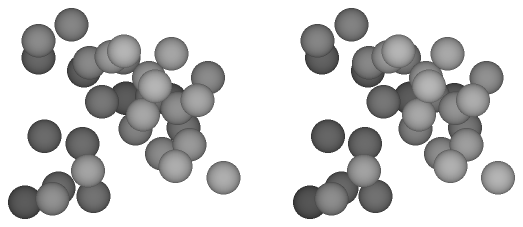
As we stated in the previous section, the cluster phase and the monomer phase should be simulated independently for the purpose of the free energy estimation. Then, we performed four stages of the MC calculations: namely, cluster stabilization process, phase transition calculation, cluster phase calculation, and monomer phase calculation. The first stage, the cluster stabilization, prepares an initial cluster for the phase transition and the cluster phase calculations. An initial configuration of this stage is chosen among distorted simple cubic, body-centered cubic, or face-centered cubic lattices by the number of particles. If the N particles do not fill the lattice, the defect(s) is (are) inserted into the lattice site(s). Starting with the initial lattice, the particles are stabilized as a cluster by the cooling MC calculation. From the initial temperature 0.20 ε/k, the system is cooled to 0.01 ε/k in 0.01 ε/k steps. We calculated at least 4 million MC steps severally for equilibration and statistics at each temperature; where we regarded one MC step into N trial movements. The second stage is the phase transition calculation. The stabilized cluster created in the first stage is heated up from 0.01 to 1.00 ε/k in 0.01 ε/k steps; where one million MC steps were calculated for equilibration and statistics at each temperature. The cluster decomposes to the monomer phase (the supersaturated vapor phase) at a certain temperature that depends on the number of particles. Thirdly, the cluster phase calculation also uses the stabilized cluster as an initial configuration and the system is heated up similarly to the second stage. However, the cluster decomposition, i.e., leaving of any particle from the N-particle cluster, is prohibited in this stage. We applied Stillinger's cluster criterion[12] for this purpose. According to the Stillinger's criterion, any two particles are connected if their center-center distance is less than a threshold distance. After each MC movement, we examined the new configuration whether all particles of the system are mutually connected. If the new configuration was not considered as a unity cluster, it was rejected before the Metropolis judgment and was not counted as the number of the MC trial. We selected the threshold distance as 1.5 σ, that has been used and examined widely,[2,3,5] and corresponds to the first minimum of the pair correlation function of the cluster (or bulk liquid) phase. The third stage obtains the superheated cluster phase above the phase transition temperature. Lastly, we performed the monomer phase calculation. Contrary to the cluster phase calculation, any connections should be refused in this stage. That is to say, all combinations of the center-center distances of the particles in the system should be longer than 1.5 σ. After each MC trials, if the new configuration had any connections, the trial was canceled. The initial lattice is similar to the cluster stabilization process, however all the interparticle distances are set to be longer than the threshold value as the monomer phase. Starting with initial temperature 1.00 ε/k, the system was cooled to 0.01 ε/k in 0.01 ε/k steps; where between 100 thousands to one million MC steps were calculated for equilibration and statistics severally at each temperature. Supercooled monomer phase is obtained in this stage. Figures 2.5-2.8 show sample snapshots of the stabilized cluster, the decomposed monomer phase in the phase transition calculation, the superheated cluster phase, and the supercooled monomer phase on 32-particle system.
|
|
2.4. Free Energy Estimation |
An interaction term of Helmholtz free energy Ae under constant volume is estimated as
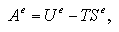 | (2.3) |
where Ue indicates an average value of interaction energy of the system, T temperature and Se an interaction term of entropy. During the MC calculation, the Ue can be easily obtained as the sum of two-body interaction energies as
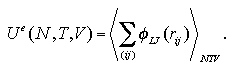 | (2.4) |
However, there are some difficulties in obtaining the interaction entropy. We estimated the interaction entropy as follows:[9]
We obtained an interaction term of the heat capacity as the numerical differentiation (eq. 2.5) or the fluctuation (eq. 2.6) of the interaction energy, besides we obtained the interaction entropy as the thermodynamic integration (eq. 2.7) of the heat capacity. Accordingly, the interaction entropy obtained in this study is a relative value that has the same starting point as the heat capacity at T0 = 0.01 ε/k.
When the Helmholtz free energies of the cluster phase Ace and monomer phase Ame under constant volume are obtained independently, the free energy change of the nucleation ΔA is regarded as
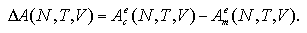 | (2.9) |
In eqs. 2.3-2.9, we neglected the ideal gas terms of internal energy, heat capacity, entropy, and Helmholtz free energy. However, if the volume is constant, the ideal gas terms are identical irrespective of the configuration of the system, and canceled by subtraction in eq. 2.9. Therefore, the neglect is adequate for the purpose of ΔA estimation.
On the other hand, Gibbs free energy is sum of the Helmholtz free energy and the product of the pressure P and the volume V:
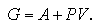 | (2.10) |
The Gibbs free energy of the nucleation ΔG should be obtained under constant pressure as
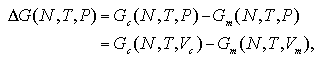 | (2.11) |
where Vc and Vm are volumes of the cluster phase and the monomer phase that present an identical pressure P. In this equation, the ideal gas terms of the each phase are not canceled because the ideal gas term of the Gibbs free energy depends on the volume. Accordingly, to obtain the ΔG on (N,T,P) domain, the ideal gas terms Gcid and Gmid should be taken into account as
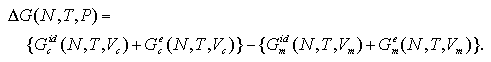 | (2.12) |
|